














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
BOTOX® Cosmetic (onabotulinumtoxinA) IMPORTANT SAFETY INFORMATION, INCLUDING BOXED WARNING
WARNING: DISTANT SPREAD OF TOXIN EFFECT
Postmarketing reports indicate that the effects of BOTOX® Cosmetic and all botulinum toxin products may spread from the area of injection to produce symptoms consistent with botulinum toxin effects. These may include asthenia, generalized muscle weakness, diplopia, ptosis, dysphagia, dysphonia, dysarthria, urinary incontinence and breathing difficulties. These symptoms have been reported hours to weeks after injection. Swallowing and breathing difficulties can be life threatening and there have been reports of death. The risk of symptoms is probably greatest in children treated for spasticity but symptoms can also occur in adults treated for spasticity and other conditions, particularly in those patients who have an underlying condition that would predispose them to these symptoms. In unapproved uses, including spasticity in children, and in approved indications, cases of spread of effect have been reported at doses comparable to those used to treat cervical dystonia and upper limb spasticity and at lower doses.
Important Information
Indications
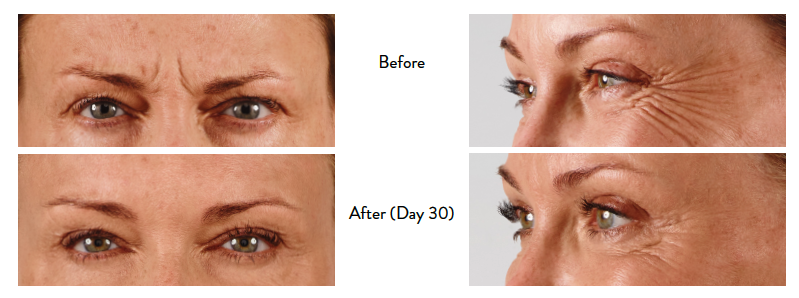
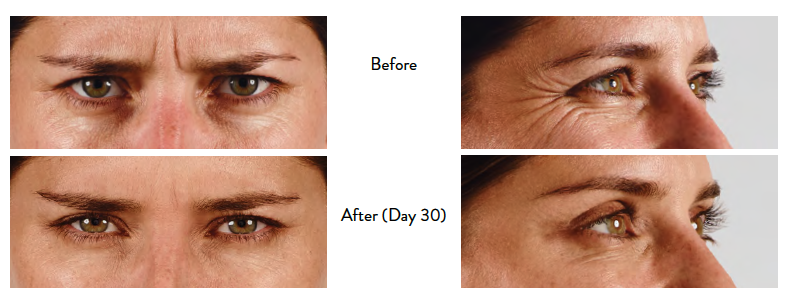
Glabellar Lines
BOTOX® Cosmetic (onabotulinumtoxinA) for injection is indicated for the temporary improvement in the appearance of moderate to severe glabellar lines associated with corrugator and/or procerus muscle activity in adult patients.
Lateral Canthal Lines
BOTOX® Cosmetic is indicated for the temporary improvement in the appearance of moderate to severe lateral canthal lines associated with orbicularis oculi activity in adult patients.
IMPORTANT SAFETY INFORMATION (continued)
CONTRAINDICATIONS
BOTOX® Cosmetic is contraindicated in the presence of infection at the proposed injection site(s) and in individuals with known hypersensitivity to any botulinum toxin preparation or to any of the components in the formulation.
WARNINGS AND PRECAUTIONS
Lack of Interchangeability between Botulinum Toxin Products
The potency units of BOTOX® Cosmetic are specific to the preparation and assay method utilized. They are not interchangeable with other preparations of botulinum toxin products and, therefore, units of biological activity of BOTOX® Cosmetic cannot be compared to nor converted into units of any other botulinum toxin products assessed with any other specific assay method.
Spread of Toxin Effect
Please refer to Boxed Warning for Distant Spread of Toxin Effect.
No definitive serious adverse event reports of distant spread of toxin effect associated with dermatologic use of BOTOX® Cosmetic at the labeled dose of 20 Units (for glabellar lines), 24 Units (for lateral canthal lines), 44 Units (for simultaneous treatment of lateral canthal lines and glabellar lines) have been reported.
Serious Adverse Reactions With Unapproved Use
Serious adverse reactions, including excessive weakness, dysphagia, and aspiration pneumonia, with some adverse reactions associated with fatal outcomes, have been reported in patients who received BOTOX® injections for unapproved uses. In these cases, the adverse reactions were not necessarily related to distant spread of toxin, but may have resulted from the administration of BOTOX® to the site of injection and/or adjacent structures. In several of the cases, patients had pre-existing dysphagia or other significant disabilities. There is insufficient information to identify factors associated with an increased risk for adverse reactions associated with the unapproved uses of BOTOX®. The safety and effectiveness of BOTOX® for unapproved uses have not been established.
Hypersensitivity Reactions
Serious and/or immediate hypersensitivity reactions have been reported. These reactions include anaphylaxis, serum sickness, urticaria, soft-tissue edema, and dyspnea. If such reactions occur, further injection of BOTOX® Cosmetic should be discontinued and appropriate medical therapy immediately instituted. One fatal case of anaphylaxis has been reported in which lidocaine was used as the diluent and, consequently, the causal agent cannot be reliably determined.
Cardiovascular System
There have been reports following administration of BOTOX® of adverse events involving the cardiovascular system, including arrhythmia and myocardial infarction, some with fatal outcomes. Some of these patients had risk factors including pre-existing cardiovascular disease. Use caution when administering to patients with pre-existing cardiovascular disease.
Pre-existing Neuromuscular Disorders
Individuals with peripheral motor neuropathic diseases, amyotrophic lateral sclerosis, or neuromuscular junction disorders (eg, myasthenia gravis or Lambert-Eaton syndrome) should be monitored when given botulinum toxin. Patients with neuromuscular disorders may be at increased risk of clinically significant effects including generalized muscle weakness, diplopia, ptosis, dysphonia, dysarthria, severe dysphagia, and respiratory compromise from onabotulinumtoxinA (see Warnings and Precautions).
Dysphagia and Breathing Difficulties
Treatment with BOTOX® and other botulinum toxin products can result in swallowing or breathing difficulties. Patients with pre-existing swallowing or breathing difficulties may be more susceptible to these complications. In most cases, this is a consequence of weakening of muscles in the area of injection that are involved in breathing or oropharyngeal muscles that control swallowing or breathing (see Boxed Warning).
Pre-existing Conditions at the Injection Site
Caution should be used when BOTOX® Cosmetic treatment is used in the presence of inflammation at the proposed injection site(s) or when excessive weakness or atrophy is present in the target muscle(s).
Human Albumin and Transmission of Viral Diseases
This product contains albumin, a derivative of human blood. Based on effective donor screening and product manufacturing processes, it carries an extremely remote risk for transmission of viral diseases. A theoretical risk for transmission of Creutzfeldt-Jakob disease (CJD) also is considered extremely remote. No cases of transmission of viral diseases or CJD have ever been identified for albumin.
ADVERSE REACTIONS
The most frequently reported adverse event following injection of BOTOX® Cosmetic for glabellar lines was eyelid ptosis (3%).
The most frequently reported adverse event following injection of BOTOX® Cosmetic for lateral canthal lines was eyelid edema (1%).
DRUG INTERACTIONS
Co-administration of BOTOX® Cosmetic and aminoglycosides or other agents interfering with neuromuscular transmission (eg, curare-like compounds) should only be performed with caution as the effect of the toxin may be potentiated. Use of anticholinergic drugs after administration of BOTOX® Cosmetic may potentiate systemic anticholinergic effects.
The effect of administering different botulinum neurotoxin products at the same time or within several months of each other is unknown. Excessive neuromuscular weakness may be exacerbated by administration of another botulinum toxin prior to the resolution of the effects of a previously administered botulinum toxin.
Excessive weakness may also be exaggerated by administration of a muscle relaxant before or after administration of BOTOX® Cosmetic.
USE IN SPECIFIC POPULATIONS
BOTOX® Cosmetic is not recommended for use in children or pregnant women. It is not known whether BOTOX® Cosmetic is excreted in human milk. Caution should be exercised when BOTOX® Cosmetic is administered to a nursing woman.
Please see BOTOX® Cosmetic full Prescribing Information including Boxed Warning and Medication Guide.
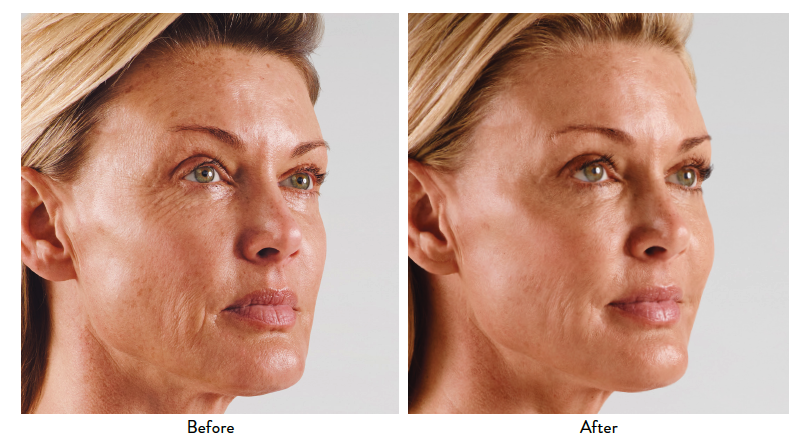
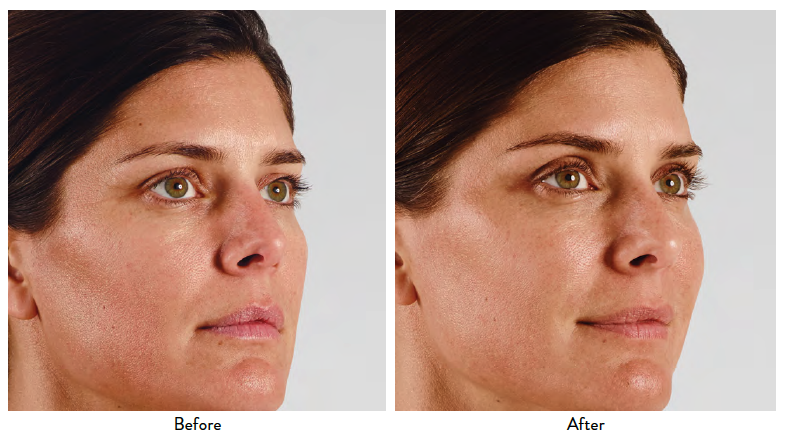
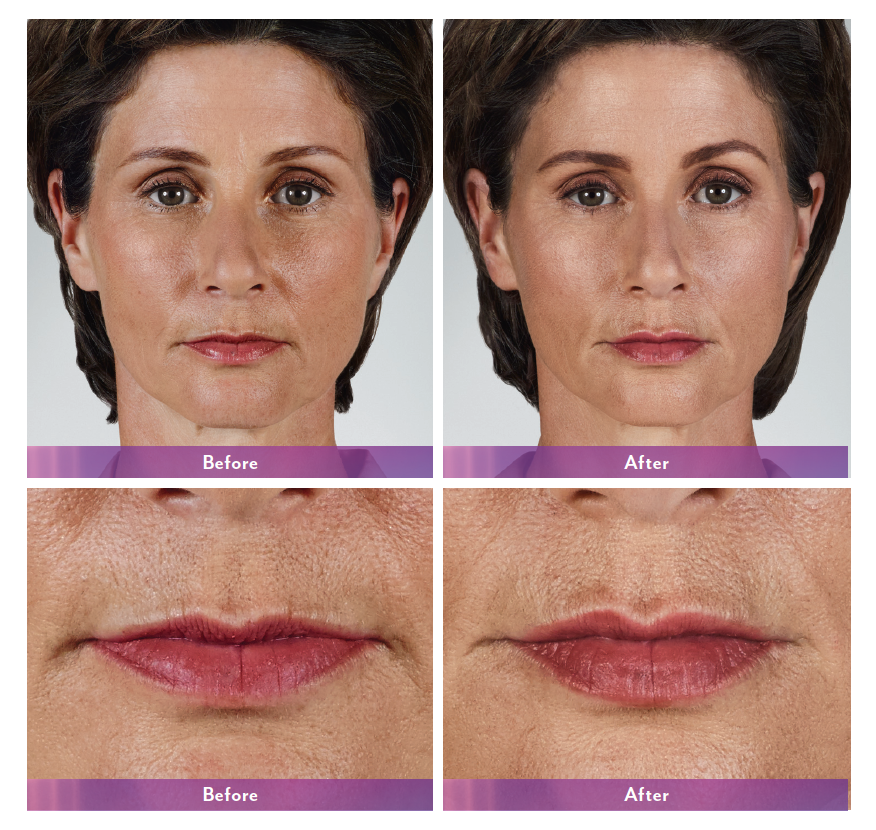
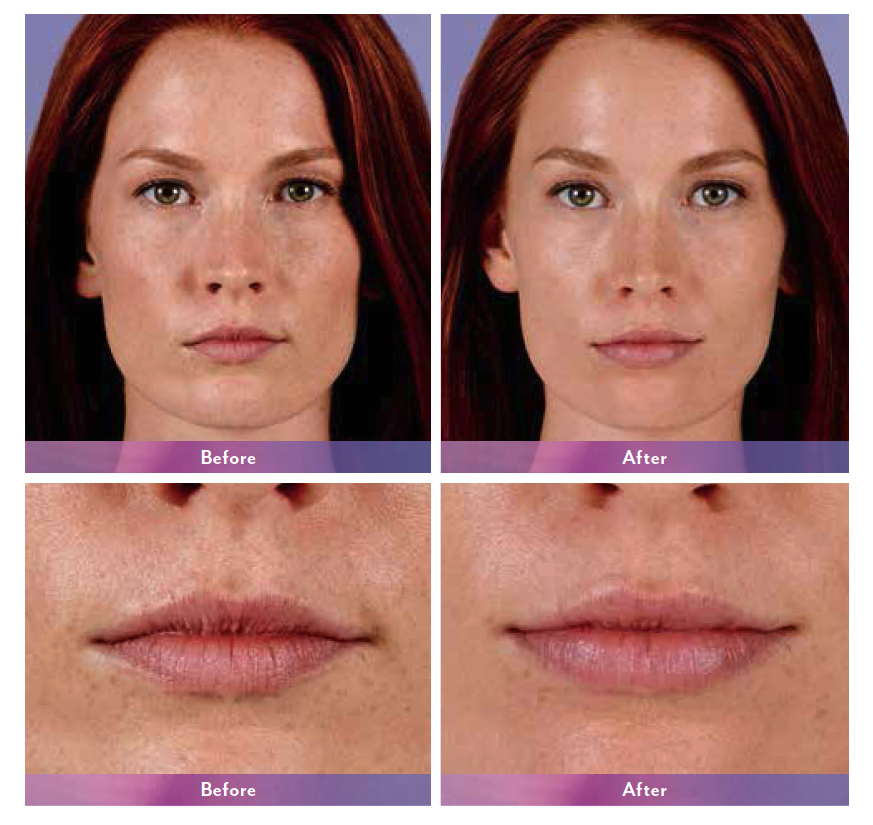
JUVÉDERM® XC, JUVÉDERM® Ultra XC, JUVÉDERM VOLUMA® XC, and JUVÉDERM VOLBELLA® XC Important Information
INDICATIONS
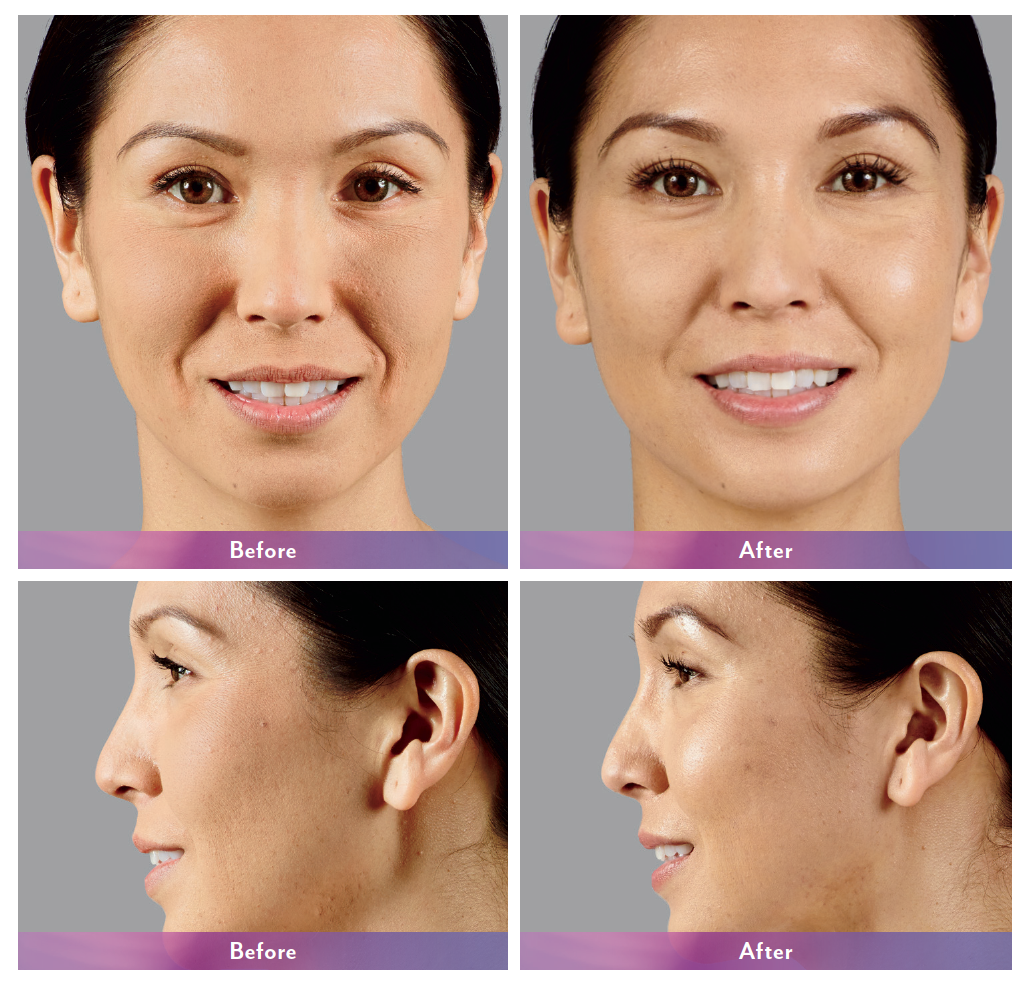
JUVÉDERM® XC injectable gels (JUVÉDERM® Ultra XC and JUVÉDERM® Ultra Plus XC) are indicated for injection into the mid-to-deep dermis for correction of moderate to severe facial wrinkles and folds (such as nasolabial folds).
JUVÉDERM® Ultra XC injectable gel is indicated for injection into the lips and perioral area for lip augmentation in adults over the age of 21.
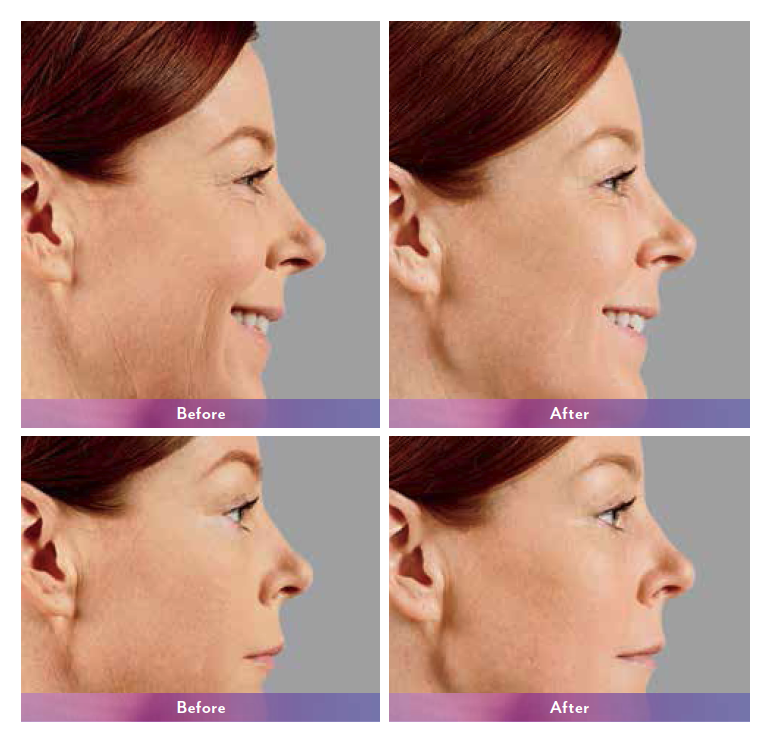
JUVÉDERM VOLUMA® XC injectable gel is indicated for deep (subcutaneous and/or supraperiosteal) injection for cheek augmentation to correct age-related volume deficit in the mid-face in adults over the age of 21.
JUVÉDERM VOLBELLA® XC injectable gel is indicated for injection into the lips for lip augmentation and for correction of perioral rhytids in adults over the age of 21.
IMPORTANT SAFETY INFORMATION
CONTRAINDICATIONS
These products should not be used in patients who have severe allergies, marked by a history of anaphylaxis or history or presence of multiple severe allergies, and should not be used in patients with a history of allergies to Gram-positive bacterial proteins or lidocaine contained in these products.
WARNINGS
- Do not inject into blood vessels. Introduction of these products into the vasculature may lead to embolization, occlusion of the vessels, ischemia, or infarction. Take extra care when injecting soft-tissue fillers; for example, inject the product slowly and apply the least amount of pressure necessary. Rare, but serious, adverse events associated with the intravascular injection of soft-tissue fillers in the face have been reported and include temporary or permanent vision impairment, blindness, cerebral ischemia or cerebral hemorrhage leading to stroke, skin necrosis, and damage to underlying facial structures. Immediately stop the injection if a patient exhibits any of the following symptoms: changes in vision, signs of a stroke, blanching of the skin, unusual pain during or shortly after the procedure. Patients should receive prompt medical attention and, possibly, evaluation by an appropriate healthcare professional specialist should an intravascular injection occur
- Product use at specific sites in which an active inflammatory process (skin eruptions such as cysts, pimples, rashes, or hives) or infection is present should be deferred until the underlying process has been controlled
PRECAUTIONS
- In order to minimize the risk of potential complications, these products should only be used by healthcare professionals who have appropriate training, experience, and knowledge of facial anatomy
- Healthcare professionals are encouraged to discuss the potential risks of soft-tissue injections with their patients prior to treatment and ensure that patients are aware of signs and symptoms of potential complications
- The safety and effectiveness for the treatment of anatomic regions other than moderate to severe facial wrinkles and folds with JUVÉDERM® Ultra XC and JUVÉDERM® Ultra Plus XC, the lips and perioral area for lip augmentation with JUVÉDERM® Ultra XC, the mid-face with JUVÉDERM VOLUMA®XC, and the lips and perioral area for lip augmentation and correction of perioral rhytids with JUVÉDERM VOLBELLA® XC have not been established in controlled clinical studies
- As with all transcutaneous procedures, dermal filler implantation carries a risk of infection. Follow standard precautions associated with injectable materials
- The safety for use during pregnancy, in breastfeeding females, and in patients with known susceptibility to keloid formation, hypertrophic scarring, and pigmentation disorders has not been studied
- The safety for use of JUVÉDERM® Ultra XC and JUVÉDERM® Ultra Plus XC in patients under 18 years, JUVÉDERM VOLUMA® XC in patients under 35 or over 65 years, and JUVÉDERM VOLBELLA® XC in patients under 22 years has not been established
- Use with caution in patients on immunosuppressive therapy
- Patients who are using products that can prolong bleeding (such as aspirin, nonsteroidal anti-inflammatory drugs, and warfarin) may experience increased bruising or bleeding at treatment sites
- If laser treatment, chemical peel, or any other procedure based on active dermal response is considered after treatment, or if the product is administered before the skin has healed completely, there is a possible risk of an inflammatory reaction at the treatment site
- Patients who experience skin injury near the site of implantation may be at a higher risk for adverse events
- The safety of JUVÉDERM VOLUMA® XC injectable gel for use in patients with very thin skin in the mid-face has not been established
- Patients may experience late onset nodules with use of dermal fillers, including JUVÉDERM VOLUMA® XC
- Patients may experience late onset adverse events with use of dermal fillers, including JUVÉDERM VOLBELLA® XC
ADVERSE EVENTS
The most commonly reported side effects for JUVÉDERM® injectable gels were temporary injection-site redness, swelling, pain/tenderness, firmness, lumps/bumps, bruising, discoloration, and itching. For JUVÉDERM VOLBELLA® XC, dryness was also reported. For JUVÉDERM® Ultra XC or JUVÉDERM® Ultra Plus XC, side effects were mostly mild or moderate in severity, with a duration of 14 days or less; for JUVÉDERM VOLUMA® XC, they were predominantly moderate in severity, with a duration of 2 to 4 weeks; and for JUVÉDERM VOLBELLA® XC, they were predominantly mild or moderate, with a duration of 30 days or less.
To report an adverse reaction with JUVÉDERM® Ultra XC, JUVÉDERM® Ultra Plus XC, JUVÉDERM VOLUMA® XC, or JUVÉDERM VOLBELLA® XC, please call Allergan at 1-800-433-8871. Please also visit JuvedermDFU.com for more information.
JUVÉDERM® Ultra XC, JUVÉDERM® Ultra Plus XC, JUVÉDERM VOLUMA® XC, and JUVÉDERM VOLBELLA® XC injectable gels are available by prescription only.
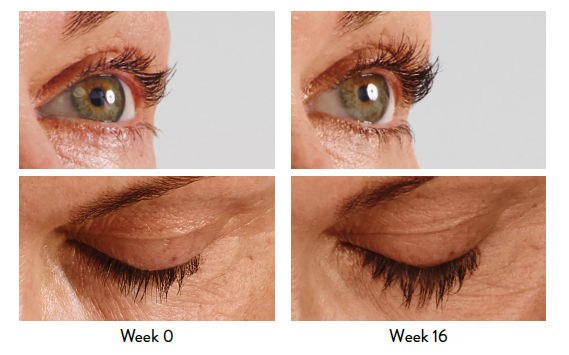
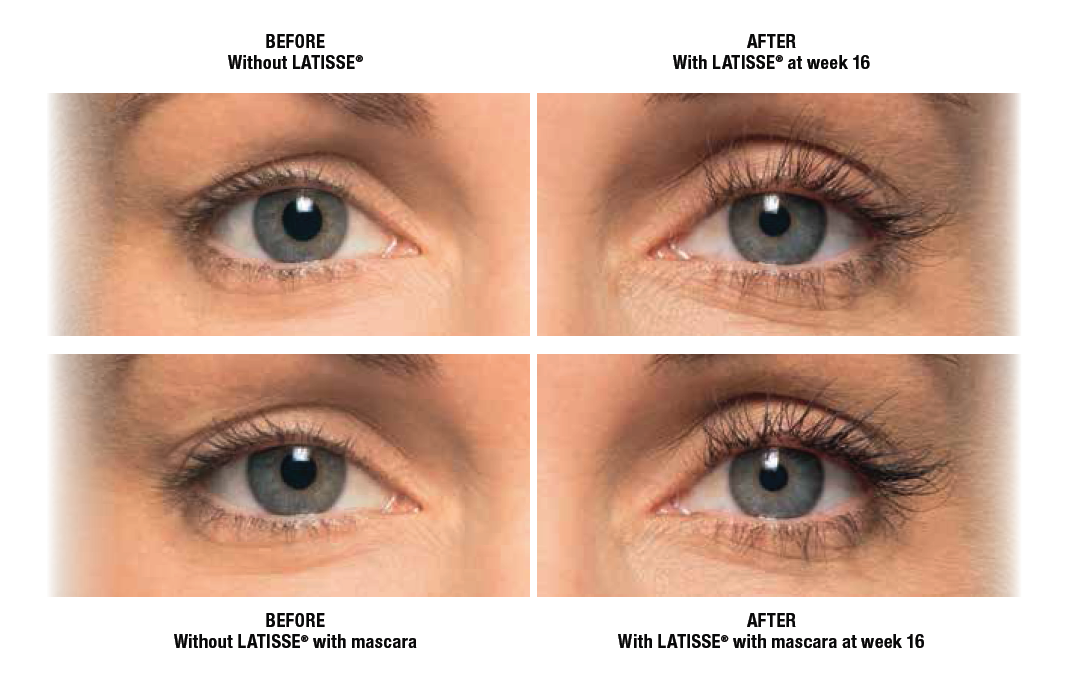
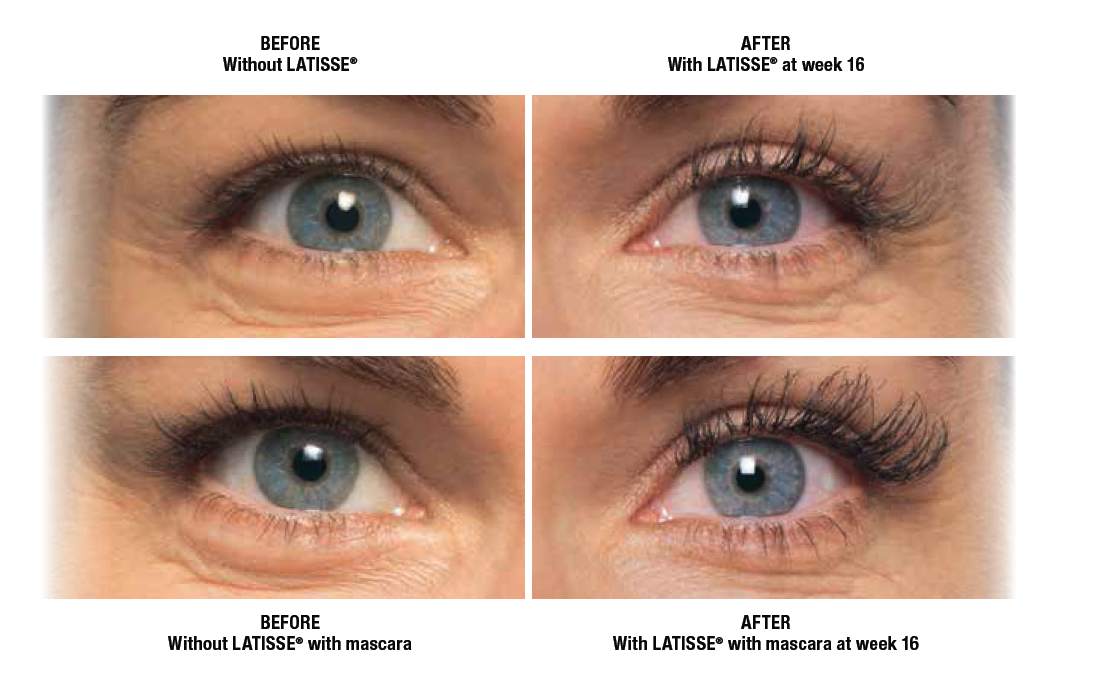
LATISSE® (bimatoprost ophthalmic solution) 0.03% Important Information
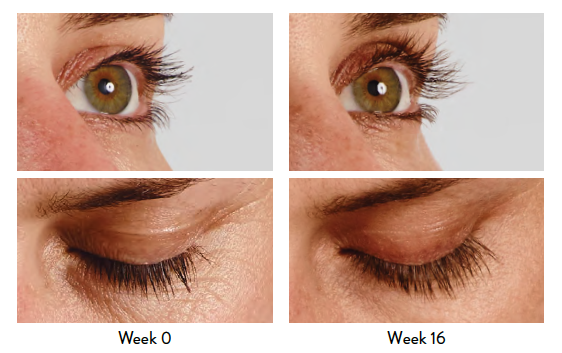
Indication
LATISSE® (bimatoprost ophthalmic solution) 0.03% is indicated to treat hypotrichosis of the eyelashes by increasing their growth, including length, thickness, and darkness.
Important Safety Information
Warnings and Precautions: In patients using LUMIGAN® (bimatoprost ophthalmic solution) or other prostaglandin analogs for the treatment of elevated intraocular pressure (IOP), the concomitant use of LATISSE® may interfere with the desired reduction in IOP. Patients using prostaglandin analogs including LUMIGAN® for IOP reduction should only use LATISSE® after consulting with their physician and should be monitored for changes to their intraocular pressure.
Increased iris pigmentation has occurred when bimatoprost solution was administered. Patients should be advised about the potential for increased brown iris pigmentation, which is likely to be permanent.
Bimatoprost has been reported to cause pigment changes (darkening) to periorbital pigmented tissues and eyelashes. The pigmentation is expected to increase as long as bimatoprost is administered, but has been reported to be reversible upon discontinuation of bimatoprost in most patients.
There is the potential for hair growth to occur in areas where LATISSE® solution comes in repeated contact with skin surfaces. Apply LATISSE® only to the skin of the upper eyelid margin at the base of the eyelashes.
LATISSE® solution should be used with caution in patients with active intraocular inflammation (eg, uveitis) because the inflammation may be exacerbated.
Adverse Reactions: The most frequently reported adverse events were eye pruritus, conjunctival hyperemia, skin hyperpigmentation, ocular irritation, dry eye symptoms, and erythema of the eyelid. These events occurred in less than 4% of patients.
Postmarketing Experience: The following reactions have been identified during postmarketing use of LATISSE® in clinical practice: eye swelling, eyelid edema, hypersensitivity (local allergic reactions), lacrimation increased, madarosis and trichorrhexis (temporary loss of a few eyelashes to loss of sections of eyelashes, and temporary eyelash breakage, respectively), periorbital and lid changes associated with a deepening of the eyelid sulcus, rash (including macular and erythematous), skin discoloration (periorbital), and vision blurred.
Please see LATISSE® full Prescribing Information.